
Reproducible research: Recording computational steps for OSeMOSYS
Overview
Exercises: 25 min
- How can we create a reproducible workflow?
- When to use scientific workflow management systems.
- Discuss pros/cons of GUI vs. manual steps vs. scripted vs. workflow tools.
- Get familiar with Snakemake.
One problem solved in different ways
The following material is adapted from a HPC Carpentry lesson
Let’s look at an example project which follows the project structure guidelines given in the previous episode. The project runs OSeMOSYS and plots a couple of charts.
To follow along, clone this repository:
$ git clone https://github.com/KTH-dESA/osemosys_workflow.git
$ git checkout simple
The example project directory listing is:
.
├── README.md
├── data
│ ├── README.md
│ └── simplicity.txt
├── env
│ ├── dag.yaml
│ ├── osemosys.yaml
│ └── plot.yaml
├── model
│ ├── LICENSE
│ └── osemosys.txt
├── processed_data
├── scripts
│ ├── osemosys_run.sh
│ └── plot_results.py
└── snakefile
In this example we wish to:
- Run a model run
- Extract some results from the
SelectedResults.csvfile and save them into separate csv files - Plot those results
Ideally, we would go on to:
- Create environments for each of the key tasks
- Run model runs in parallel and scale them up for running on a cluster
The Problem
Example (for one model run only) - let us test this out:
$ glpsol -d data/simplicity.txt -m model/osemosys.txt -o processed_data/results.sol
$ ???
$ python scripts/plot_results.py processed_data/total_annual_capacity.csv results/total_annual_capacity.pdf
$ ???
$ python scripts/plot_results.py processed_data/tid_demand.csv results/tid_demand.pdf
Can you relate? Are you using similar setups in your research?
This was for one model run - how about 3 model runs? How about 3000 model runs?
Discussion
Discuss the pros and cons of this approach. Is it reproducible? Does it scale to hundreds of model runs? Can it be automated? What if you modify only one data file and do not wish to rerun the pipeline for all datafiles again?
Exercise
What commands are required to replace the
???in the second line? Think back to the first workshop on the shell…
Solution 1: Script
Let’s express it more compactly with a shell script (Bash). Let’s call it run_analysis.sh:
#!/usr/bin/env bash
glpsol -d data/simplicity.txt -m model/osemosys.txt -o processed_data/results.sol
head -n 326 processed_data/SelectedResults.csv | tail -n 29 > processed_data/total_annual_capacity.csv
python scripts/plot_results.py processed_data/total_annual_capacity.csv results/total_annual_capacity.pdf
head -n 33 processed_data/SelectedResults.csv | tail -n 22 > processed_data/tid_demand.csv
python scripts/plot_results.py processed_data/tid_demand.csv results/tid_demand.pdf
We can run it with:
$ run_analysis.sh
This is still imperative style: we tell the script to run these steps in precisely this order.
Exercise/discussion
Discuss the pros and cons of this approach. Is it reproducible? Does it scale to hundreds of model runs? Can it be automated? What if you modify only one data file and do not wish to rerun the pipeline for all datafiles again?
Solution 2: Using GNU Make
- A tool from the 70s often used to build software.
- Uses specific syntax that the user writes in a Makefile.
- Makefile specifies how to build targets from their dependencies.
- Observe that we use wildcards instead of explicit model run names.
It contains rules that relate targets to dependencies and commands:
# rule (mind the tab)
target: dependencies
command(s)
We can think of it as follows:
outputs: inputs
command(s)
Make uses declarative style: we describe dependencies but we let Make figure out the series of steps to produce results (targets). Fun fact: Excel is also declarative, not imperative.
Solution 3: Using Snakemake
Switch to the simple branch:
git checkout simple
First study a simple Snakefile:
rule all:
input: "processed_data/SelectedResults.csv"
rule solve:
input: data="data/simplicity.txt", model="model/osemosys.txt"
output: results="processed_data/results.sol", default="processed_data/SelectedResults.csv"
log: "processed_data/glpsol.log"
shell:
"glpsol -d {input.data} -m {input.model} -o {output.results} > {log}"
rule clean:
shell:
"rm -f processed_data/*.sol processed_data/*.csv processed_data/*.log"
Snakemake uses the declarative style:
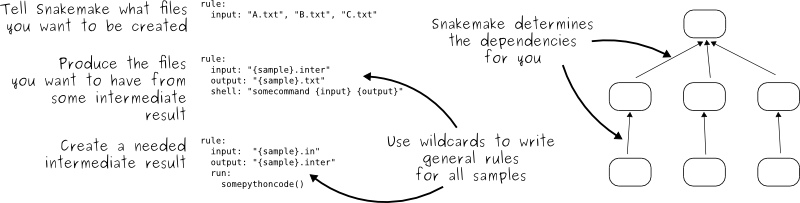
Try it out:
$ snakemake clean
$ snakemake
Try running snakemake again and observe that and discuss why it refused to rerun all steps:
$ snakemake
Building DAG of jobs...
Nothing to be done.
Make a modification to the data file and run snakemake again and discuss
what you see. One way to modify files is to use the touch command which will
only update its timestamp:
$ touch data/simplicity.txt
$ snakemake
You can try a dry run with the -n flag, if you’re not sure what’s going to be built:
$ snakemake clean
$ snakemake -n
Exercise: extending the simple example
Create snakemake rules to extract our data tables and write them to a csv file.
Hint:
head -n 326 processed_data/SelectedResults.csv | tail -n 29 > processed_data/total_annual_capacity.csv
head -n 33 processed_data/SelectedResults.csv | tail -n 22 > processed_data/tid_demand.csv
Solution
rule extract_tid_demand:
input: "processed_data/SelectedResults.csv"
output: "processed_data/tid_demand.csv"
shell:
"head -n 33 {input} | tail -n 22 > {output}"
Exercise: plotting the csv files
Create snakemake rules to generate the plots:
Hint:
python scripts/plot_results.py processed_data/total_annual_capacity.csv results/total_annual_capacity.pdf
python scripts/plot_results.py processed_data/tid_demand.csv results/tid_demand.pdf
After creating your new rules, try running snakemake. Why does nothing happen?
Creating targets
The first command in your snakemake file should contain a list of targets - the final elements you want to produce.
RESULTS = ['tid_demand', 'total_annual_capacity']
rule all:
input: expand("processed_data/{x}.pdf", x=RESULTS)
message: "Running pipeline to generate the files '{input}'"
Generalising Snakemake Rules
Switch to the general branch:
$ git checkout general
The rules we made in the previous exercise were very very similar to one another.
This is a common coding “smell” and should be a warning to you to refactor your code.
rule plot:
input: "processed_data/{result}.csv"
output: "processed_data/{result}.pdf"
message: "Generating plot using '{input}' and writing to '{output}'"
shell:
"python scripts/plot_results.py {input} {output}"
Environments
If you install snakemake with conda, you can define conda environments per rule.
rule solve:
input: data="data/simplicity.txt", model="model/osemosys.txt"
output: results="processed_data/results.sol", default="processed_data/SelectedResults.csv"
log: "processed_data/glpsol.log"
conda: "env/osemosys.yaml"
shell:
"glpsol -d {input.data} -m {input.model} -o {output.results} > {log}"
The env/osemosys.yaml file:
channels:
- conda-forge
- defaults
dependencies:
- glpk
Then, running snakemake with the --use-conda flag:
$ snakemake --use-conda --cores 2
Building DAG of jobs...
Using shell: /usr/local/bin/bash
Provided cores: 2
Rules claiming more threads will be scaled down.
Job counts:
count jobs
1 all
1 extract_tid_demand
1 extract_total_annual_capacity
2 plot
1 solve
6
[Wed Sep 11 22:53:50 2019]
rule solve:
input: data/simplicity.txt, model/osemosys.txt
output: processed_data/results.sol, processed_data/SelectedResults.csv
log: processed_data/glpsol.log
jobid: 5
Activating conda environment: /Users/wusher/repository/osemosys_snakemake/.snakemake/conda/6fbfeaaf
[Wed Sep 11 22:54:09 2019]
Finished job 5.
1 of 6 steps (17%) done
Running models runs in parallel
git checkout model_runs
We need to wrap OSeMOSYS in a little shell script to allow it to run in parallel. This allows us to change the destination of the results file which is written out:
#!/usr/bin/env bash
MODELRUN=$1
RESULTS="processed_data\/$MODELRUN\/SelectedResults.csv"
mkdir processed_data/$MODELRUN
cat model/osemosys.txt > processed_data/$MODELRUN/osemosys.txt
sed -i '' "s/FILEPATH/$RESULTS/g" processed_data/$MODELRUN/osemosys.txt
You can choose as many cores as your laptop will cope with:
snakemake --use-conda --cores 8
Why Snakemake?
- Gentle learning curve.
- Free, open-source, and installs easily via conda or pip.
- Cross-platform (Windows, MacOS, Linux) and compatible with all HPC schedulers
- same workflow works without modification and scales appropriately whether on a laptop or cluster.
- Heavily used in bioinformatics, but is completely general.
Snakemake vs. Make
- Workflows defined in Python scripts extended by declarative code to define rules:
- anything that can be done in Python can be done with Snakemake
- rules work much like in GNU Make (can call any commands/executables)
- Possible to define isolated software environments per rule.
- Also possible to run workflows in Docker or Singularity containers.
- Workflows can be pushed out to run on a cluster or in the cloud without modifications to scale up.
Visualizing the workflow
If you’re using the OSeMOSYS example, try running snakemake plot_dag.
We can visualize the directed acyclic graph (DAG) of our current Snakefile
using the --dag option, which will output the DAG in dot language (a
plain-text format for describing graphs used by Graphviz software,
which can be installed by conda install graphviz)
$ snakemake --dag | dot -Tpng > dag.png
Rules that have yet to be completed are indicated with solid outlines, while already completed rules are indicated with dashed outlines.
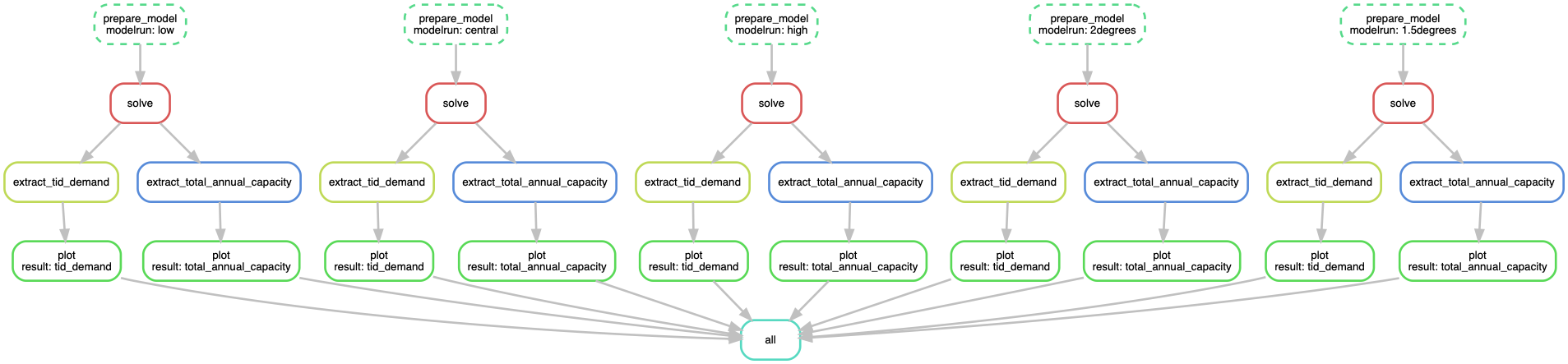
Exercise/discussion
Discuss the pros and cons of this approach. Is it reproducible? Does it scale to hundreds of model runs? Can it be automated?
More Snakemake goodies
- Archive a workflow into a tarball:
$ snakemake --archive my-workflow.tar.gz - Snakemake has an experimental GUI feature which can be invoked by:
$ snakemake --gui - Isolated software environments per rule using conda. Invoke by
snakemake --use-conda. Example:rule NAME: input: "table.txt" output: "plots/myplot.pdf" conda: "envs/ggplot.yaml" script: "scripts/plot-stuff.R" - It is possible to address and offload to non-CPU resources:
$ snakemake clean $ snakemake -j 4 --resources gpu=1 - Transferring your workflow to a cluster:
$ snakemake --archive myworkflow.tar.gz $ scp myworkflow.tar.gz <some-cluster> $ ssh <some-cluster> $ tar zxf myworkflow.tar.gz $ cd myworkflow $ snakemake -n --use-conda - Interoperability with Slurm:
{ "__default__": { "account": "a_slurm_submission_account", "mem": "1G", "time": "0:5:0" }, "count_words": { "time": "0:10:0", "mem": "2G" } }The workflow can now be executed by:
$ snakemake -j 100 --cluster-config cluster.json --cluster "sbatch -A {cluster.account} --mem={cluster.mem} -t {cluster.time} -c {threads}"Note that in this case
-jdoes not correspond to the number of cores used, instead it represents the maximum number of jobs that Snakemake is allowed to have submitted at the same time. The--cluster-configflag specifies the config file for the particular cluster, and the--clusterflag specifies the command used to submit jobs on the particular cluster. - Jobs can be run in containers. Execute with
snakemake --use-singularity. Example:rule NAME: input: "table.txt" output: "plots/myplot.pdf" singularity: "docker://joseespinosa/docker-r-ggplot2" script: "scripts/plot-stuff.R" - There is a lot more: snakemake.readthedocs.io.
Comparison and summary
- GUIs may or may not be reproducible.
- Some GUIs can be automated, many cannot.
- Typing the same series of commands for 100 similar inputs is tedious and error prone.
- Imperative scripts are reproducible and great for automation.
- Declarative workflows such as Snakemake are great for longer multi-step analyses.
- Declarative workflows are often easy to parallelize without you changing anything.
- With declarative workflows it is no problem to add/change things late in the project.
- Many specialized frameworks exist.
Key points
Preserve the steps for re-generating published results.
Hundreds of workflow management tools exist.
Make and Snakemake are a comparatively simple and lightweight options to create transferable and scalable data analyses.
Sometimes a script is enough.